Covid, FDA Disavows CDC and Promotes RT-PCR Swabs despite “Risks of False Positives”. Amazon’s Tests Sold Out due to Omicron Spreading

Amazon wins variant bet:
The RT-PCR Swabs are already sold out
They was approved by FDA a fews days ago
For domestic use but not only
by Fabio Giuseppe Carlo Carisio
July 21, 2021 was a historic date for real-time RT PCR swabs that even their inventor Kary Banks Mullis, Nobel Prize in Chemistry in 1993 for the discovery of the Polymerase Chain Reaction (PCR) technique, had recommended not to use for the diagnosis of pathologies.
On that day the Centers of Diseases Control and Prevention (CDC), the highest health authorities of every American state, advised the US laboratories that they would request the withdrawal of the Emergency Use Authorization (EUA) of this type of test because not specific enough to distinguish Covid-19 from flu.
Fact-checkers of the mainstream media, funded by Bill Gates to protect the international narrative on the pandemic and the contagion alarm aimed at justifying the Big Pharma business on vaccines, climbed on the windows of the skyscrapers in order to weaken the scope of the CDC document that called on laboratories to find new diagnostic methods, including multiplex IVD in vitro tests.
In the USA the Food and Drug Administration has worked hard to defend those tampons that represent the only proof of the surge in contagions of the Omicron variant. in the USA, the United Kingdom and the European Union and the relaunch of a new season of international restrictions aimed at new boosters of experimental gene sera (except in North America where Comirnaty is already definitively approved for the over 18s, albeit amid bitter controversy) .
In fact, in the autumn, the FDA released multiple EUAs to diagnostic kits of different types ranging from the classic and unreliable RT PCR, legitimized only for serial checks with a curious “umbrella” provision, to the sophisticated multiplex BioFire Respiratory Panel 2.1 (RP2.1 ) of Biofire Diagnostics, LLC of Salt Lake City, Utah, capable of identifying 5 types of coronaviruses (including SARS-Cov-2 of course), 7 types of influenza and 7 other viral infections of the respiratory tract.
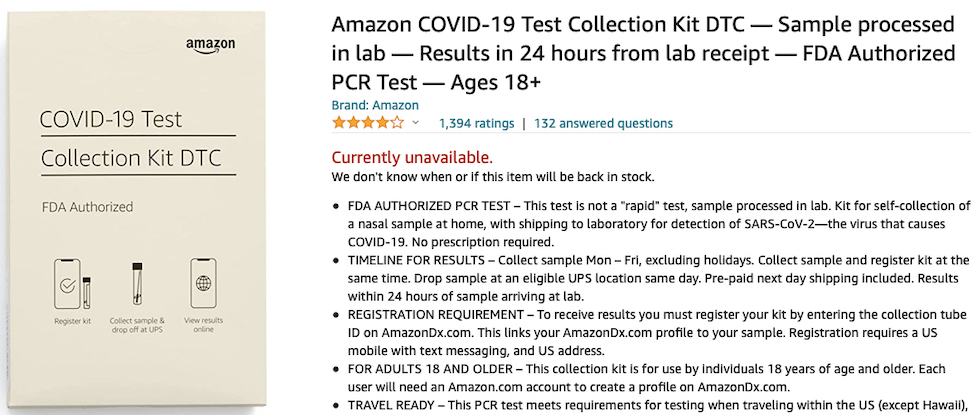
Up to the authorization of the Amazon Multi-Target SARS-CoV-2 Real-Time RT-PCR DTC domestic test created by STS Lab Holdco, a subsidiary of Amazon.com Services LLC, sold out these days on the platform of the largest worldwide online retailer.
Approved for the exclusive use of Amazon workers in March 2021, since August this ‘multi-target’ swab has also been authorized for individual tests at home by the EUA no. 210480 reconfirmed on December 17, 2021 by the FDA with four broader authorizations: not just for domestic use anymore. From emergency to colossal business, the step is always short for the Big Tech giants …
https://www.gospanews.net/2021/07/03/tamponi-covid-19-assai-inaffidabili-studi-scientifici-di-oxford-academic-e-the-lancet-nascosti-dal-mainstream-ma-citati-in-una-sentenza/
Beyond the complex bureaucratic aspects, which we will analyze in detail, the diagnostic value of RT PCR swabs, considered unreliable in 97% of cases by 3 scientific studies published in specialist journals and the historic ruling of the Lisbon Court of Appeal, however, remains uncertain as the FDA itself writes in its official documents in notes A, B, C, D, E and F of the “Limiting Statement (vi)” contextual to the approval of the super buffer BioFire Respiratory Panel 2.1.
«A negative tat does not preclude the possibility of infection. The test results should be interpreted in conjunction with other clinical and laboratory data available to the clinician; There is a risk of incorrect results due to the presence of nucleic acid sequence variants in the targeted pathogens; That positive and negative predictive values are highly dependent on prevalence; Accurate results are dependent on adequate specimen collection, transport, storage, and processing. Failure to observe proper procedures in any one of these steps can lead to incorrect results; and When applicable (e.g., recommended by the Centers for Disease Control and Prevention, by current well-accepted clinical guidelines, or by published peer-reviewed literature), that the clinical performance may be affected by testing a specific clinical subpopulation or for a specific claimed specimen type» wrote FDA from Silver Spring (MD) to Dr. Kristen Kanack, Senior Vice President, Regulatory and Clinical Affairs at BioFire.
Furthermore, in all the Fact Sheets of the RT PCR tests approved by the FDA between September and December, after the request for withdrawal of the CDC, there is still mention of the risk that nasal swabs detect “false positives” and some laboratory even warns users about serious risks deriving from this circumstance: isolation from relatives and friends, reduction of the possibility of work and prescription of treatment or therapy.
THE BIO-FIRE MULTIPLEX SWAB IS NOT INFALLIBLE
«The Center for Devices and Radiological Health (CDRH) of the Food and Drug Administration (FDA) has completed its review of your De Novo request for classification of the BioFire Respiratory Panel 2.1 (RP2.1), a prescription device with the following indications for use. The BioFire Respiratory Panel 2.1 (RP2.1) is a PCR-based multiplexed nucleic acid test intended for use with the BioFire FilmArray 2.0 or BioFire FilmArray Torch systems for the simultaneous qualitative detection and identification of multiple respiratory viral and bacterial nucleic acids in nasopharyngeal swabs (NPS) obtained from individuals suspected of respiratory tract infections, including COVID-19» we can reads in the FDA answer to the laboratory in relation to its multitarget test as an in vitro diagnostic device.
The following organism types and subtypes are identified using the BioFire RP2.1: Adenovirus, Coronavirus 229E, Coronavirus HKU1, Coronavirus NL63, Coronavirus OC43, Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV-2), Human Metapneumovirus, Human Rhinovirus/Enterovirus, Influenza A, including subtypes H1, H1-2009, and H3, Influenza B, Parainfluenza Virus 1, Parainfluenza Virus 2, Parainfluenza Virus 3, Parainfluenza Virus 4, Respiratory Syncytial Virus, Bordetella parapertussis (IS1001), Bordetella pertussis (ptxP), Chlamydia pneumoniae, and Mycoplasma pneumoniae.
«The BioFire Respiratory Panel 2.1 (RP2.1) was reviewed under the De Novo premarket review pathway, a regulatory pathway for low-to-moderate-risk devices of a new type. Along with this De Novo authorization, the FDA is establishing criteria, called special controls, that define the requirements related to labeling and performance testing» reads on the official FDA website that mentions the authorization of March 17, 2021.
When met, the special controls, in combination with general controls, provide a reasonable assurance of safety and effectiveness for tests of this type. This action also creates a new regulatory classification, which means that subsequent devices of the same type with the same intended use may go through the FDA’s 510(k) pathway, whereby devices can obtain clearance by demonstrating substantial equivalence to a predicate device.
Authorized Molecular Diagnostic Tests for SARS-CoV-2 are assigned the QJR product code. Authorized Home Collection Kits are assigned the QLW product code. Authorized EUAs for Multi-analyte Respiratory Panel Tests are assigned the QLT product code.
All this suggests a great and complete reliability of this multiplex test. But various critical issues emerge in the authorization letter, already highlighted in the past also in Italy.

«Nucleic acids from the respiratory viral and bacterial organisms identified by this test are generally detectable in NPS specimens during the acute phase of infection. The detection and identification of specific viral and bacterial nucleic acids from individuals exhibiting signs and/or symptoms of respiratory infection is indicative of the presence of the identified microorganism and aids in the diagnosis of respiratory infection if used in conjunction with other clinical and epidemiological information. The results of this test should not be used as the sole basis for diagnosis, treatment, or other patient management decisions».
The FDA’s CDRH unequivocally states in its letter to BioFire, adding: «Negative results in the setting of a respiratory illness may be due to infection with pathogens that are not detected by this test, or lower respiratory tract infection that may not be detected by an NPS specimen. Positive results do not rule out coinfection with other organisms. The agent(s) detected by the BioFire RP2.1 may not be the definite cause of disease. Additional laboratory testing (e.g. bacterial and viral culture, immunofluorescence, and radiography) may be necessary when evaluating a patient with possible respiratory tract infection».
https://www.gospanews.net/2021/12/07/tamponi-pcr-alterati-in-laboratori-partner-di-big-pharma-dei-vaccini-denuncia-di-medici-e-vittime-sui-test-ritirati-dai-cdc-usa/
The device «is intended to aid in the diagnosis of respiratory infection in conjunction with other clinical, epidemiologic, and laboratory data or other risk factors». For this in its protocol the FDA requires «a detailed device description, including device components» including «limiting factors of the device (e.g., saturation level of hybridization and maximum amplification and detection cycle number, etc.)».
“UMBRELLA” MEASURE FOR LESS RELIABLE RT-PCR TESTS
As we have in a previous investigation based on the complaint of an American association of doctors and victims of adverse reactions from vaccines, the alteration of the amplification cycles represented an element capable of producing “false positives”.
In the legal action against the US Department of Health, the complainants had shown all tests above 40 amplification cycles (including that of the CDCs themselves), considered an unreliable threshold because it was capable of identifying the positivity of any bacterium even by the academic virologist. Giorgio Palù, later appointed president of the Italian Medicines Agency.
That is why, following the request for withdrawal of RT PCR tests formulated by the CDC, «On November 15, 2021, the FDA issued an umbrella EUA for certain RT-PCR molecular-based tests, developed by laboratories, for detection of nucleic acid from SARS-CoV-2 from anterior nasal respiratory specimens for use as part of a serial testing program, that meet certain criteria for eligibility specified in the EUA. Under this EUA, authorized tests can be used with individual or pooled anterior nasal specimens for testing individuals, including individuals without symptoms or other epidemiological reasons to suspect COVID-19, when tested at least once per week».
Therefore they were considered valid only as a rough sampling if carried out «at regular intervals as part of a testing program implemented by schools, workplaces, or community groups».
On September 23, 2021, the FDA revised the EUAs of certain molecular, antigen, and serology tests to establish additional Conditions of Authorization in response to the continued emergence of new variants of SARS-CoV-2.
The FDA has determined that establishing additional conditions is necessary to mitigate the potential risk of false negative results due to either decreased sensitivity or non-reactivity associated with SARS-CoV-2 viral mutations. «The developers of authorized tests that are within the scope of the revision are now required to routinely monitor emerging viral mutations and their potential impact on the performance of the authorized SARS-CoV-2 test».
However, not the slightest hint is made of the exact opposite risk: that is, the processing of results with false positives. But all the labs report this in their Fact Sheets, even the manufacturer of the Amazon test.
THE RISKS OF FALSE POSITIVES WITH THE “NEW” RT-PCR
The UCSD EXCITE COVID-19 Test is authorized for use with anterior nasal swab specimens collected from individuals when determined to be appropriate by a healthcare provider including individuals without symptoms or other reasons to suspect COVID-19. It is produced by UCSD Biomedical Sciences of La Jolla, CA, which is very thoughtful in defining the benefits and risks of its test.
«A positive test result for COVID-19 indicates that RNA from SARS-CoV-2 was detected, and therefore the patient is infected with the virus and presumed to be contagious. Laboratory test results should always be considered in the context of clinical observations and epidemiological data (such as local prevalence rates and current outbreak/epicenter locations) in making a final diagnosis and patient management decisions. Patient management should be made by a healthcare provider and follow current CDC guidelines» writes the diagnostic laboratory that obtained the FDA emergency clearance on December 17, 2021.
«The UCSD EXCITE COVID-19 Test has been designed to minimize the likelihood of false positive test results. However, it is still possible that this test can give a false positive result, even when used in locations where the prevalence is below 5%».
But UCSD Biomedical Sciences is keen to warn users: «In the event of a false positive result, risks to patients could include the following: a recommendation for isolation of the patient, monitoring of household or other close contacts for symptoms, patient isolation that might limit contact with family or friends and may increase contact with other potentially COVID-19 patients, limits in the ability to work, delayed diagnosis and treatment for the true infection causing the symptoms, unnecessary prescription of a treatment or therapy, or other unintended adverse effects».
A similar warning comes from the George Washington University Public Health Laboratory in Washington, DC, on their authorized PCR swab on December 3, 2021: «Positive results are indicative of the presence of SARS-CoV-2 RNA; clinical correlation with patient history and other diagnostic information is necessary to determine patient infection status. Positive results do not rule out bacterial infection or co-infection with other viruses. The agent detected may not be the definite cause of disease. Laboratories within the United States and its territories are required to report all positive results to the appropriate public health authorities».
More than 2 million Ellume Covid-19 Home Tests recalled due to False Positives
And it is also similar to that of the diagnostic company STS Lab Holdco which produces the Amazon Multi-Target SARS-CoV-2 Real-Time RT-PCR DTC Test, a domestic nasopharyngeal swab to be performed by autonomously taking the biological substance and then sending it to the laboratories which give an answer in 24 hours, but subsequently authorized on December 17, 2021 with 4 different EUAs also for use not only at home.
«There is a small possibility that this test can give a positive result that is wrong (a false positive result) particularly when used in a population without many cases of COVID-19 infection» reads the information from the Amazon laboratory.
https://www.gospanews.net/2021/12/28/wuhan-gates-43-omicron-variante-per-la-quarta-dose-di-vaccini-altra-mutazione-o-nuovo-genotipo-del-sars-cov-2-da-laboratorio/
The same company then rightly specifies a particular valid for all RT PCR swabs: «This test is not yet approved or cleared by the United States FDA. FDA may issue an Emergency Use Authorization (EUA) when certain criteria are met, which includes that there are no adequate, approved, available alternatives. The EUA for this test is supported by the Secretary of Health and Human Service’s (HHS’s) declaration that circumstances exist to justify the emergency use of in vitro diagnostics for the detection and/or diagnosis of the virus that causes COVID-19».
AMAZON’S LUCKY BET ON OMICRON
«Amazon has limited sales of its at-home Covid-19 test kits amid a surge in demand tied to fears around the fast-spreading omicron variant» wrote Annie Palmer on the CNBC television network site. But today we did a verification on Amazon and the RT PCR test is temporarily no longer available.
«There have been widespread reports of long lines at Covid testing sites, while some pharmacies’ shelves of at-home Covid tests have become bare. In response to the surge, Walgreens and CVS Health said Tuesday they were limiting how many at-home Covid tests customers can purchase both in-store and online» wrote the American TV reporter.
An Amazon spokesperson told CNBC that the company is experiencing inventory shortages of some Covid tests due to increased demand. The company is working to secure additional Covid test inventory from its selling partners, the spokesperson said.
As of Wednesday, Amazon has limited the number of purchases of its own at-home PCR test kit to 10 units per shopper. Meanwhile, tests available from other vendors on its marketplace were also capped. The top-selling at-home Covid test, made by California-based iHealth Labs, was limited to five tests per person.
«It’s up to third-party sellers and vendors to set their own purchase limits, the Amazon spokesperson said. Amazon didn’t respond to questions asking when it began experiencing test shortages. The scarcity of tests comes as President Joe Biden said Tuesday the government will purchase 500 million at-home tests that Americans can order online for free. Delivery of the kits will begin next month» added CNBC journalist.
But certainly no health authority, intrigued in the colossal business of anti-Covid vaccines, will be responsible for verifying how many are symptomatic and asymptomatic, or potentially all false positives …
Only in Tanzania had RT-PCR swabs been tested with the surprising result that a papaya and a goat had found positive for Covid-19.
By a curious coincidence, they were donated to the African WHO by the foundation of the owner of Alibaba, the leading e-Commerce platform in Asia which, thanks to the lockdowns imposed for months on many businesses around the world, significantly increased its turnover. Just like Amazon …
Fabio Giuseppe Carlo Carisio
© COPYRIGHT GOSPA NEWS
prohibition of reproduction without authorization
follow Gospa News in the Telegram
MAIN SOURCES
GOSPA NEWS – WUHAN-GATES REPORTS
GOSPA NEWS – INQUIRIES CORONA VIRUS
FDA – In Vitro Diagnostics EUAs – Molecular Diagnostic Tests for SARS-CoV-2
https://www.gospanews.net/en/2021/12/12/covid-19-is-a-designed-bioweapon-with-a-toxic-structure-that-may-be-replicated-in-vaccines-us-army-retired-colonel-said/



2 pensieri su “Covid, FDA Disavows CDC and Promotes RT-PCR Swabs despite “Risks of False Positives”. Amazon’s Tests Sold Out due to Omicron Spreading”